Package การวินิจฉัยโรคโลหิตจางธาลัสซีเมีย
ทำไมต้องตรวจธาลัสซีเมียกับเรา?
โรคโลหิตจางธาลัสซีเมียในประเทศไทยเกิดจากการกลายพันธุ์ที่หลากหลายต้องใช้ผู้เชี่ยวชาญที่มีประสบการณ์ในการตรวจและแปลผลคนไทยทุกคนมีภาวะเสี่ยงต่อพาหะโรคโลหิตจางธาลัสซีเมียมากกว่า 40% โดยเป้าหมายของเรา คือต้องการให้คนไทยทุกคนได้รับบริการตรวจวินิจฉัยโรคโลหิตจางธาลัสซีเมีย แบบครอบคลุมและมาตรฐานในระดับสากลด้วย บริการการตรวจวินิจฉัยที่ครอบคลุมที่สุดในประเทศไทยและภูมิภาคเอเชียตะวันออกเฉียงใต้
โดยแบ่งการบริการออกเป็น 4 Package หลัก
1. Basic Prevention
- ครอบคลุมพาหะชนิดรุนแรง ∼85%
- เหมาะสำหรับผู้ที่ต้องการควบคุมป้องกันธาลัสซีเมียชนิดรุนแรง 3 ชนิด ตามนโยบายของกระทรวงสาธารณสุข (โรคเบต้าธาลัสซีเมียที่รุนแรง โรคเบต้าธาลัสซีเมียฮีโมโกลบินอี และโรคอัลฟาธาลัสซีเมียที่ทารกเสียชีวิตในครรภ์ (ไฮดรอป์)
การตรวจประกอบด้วย
- การตรวจความสมบูรณ์ของเม็ดเลือด CBC
- การหาชนิดและปริมาณ ฮีโมโกลบินด้วยวิธี Capillary Electrophoresis (CE)
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการขาดหายของยีน แบบ
ที่พบบ่อยคือ ชนิด SEA และ THAI
2. Comprehensive Prevention
- ครอบคลุมการกลายพันธุ์ที่พบบ่อย ∼90%
- สามารถควบคุมป้องกันโรคธาลัสซีเมียชนิดที่รุนแรงข้างต้นและโรคอัลฟ่าธาลัสซีเมีย ฮีโมโกลบิน เอชได้
การตรวจประกอบด้วย
- การตรวจความสมบูรณ์ของเม็ดเลือด CBC
- การหาชนิดและปริมาณ ฮีโมโกลบินด้วยวิธี Capillary Electrophoresis (CE)
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการขาดหายของยีน 7 แบบ ได้แก่ SEA, THAI, 3.7, 4.2, MED, FIL, 20.5
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการเปลี่ยนลำดับเบสใน DNA 6 แบบ ได้แก่ฮีโมโกลบิน Constant Spring, ฮีโมโกลบิน Pakse, ฮีโมโกลบิน Quang Sze, การกลายพันธุ์ชนิด initiation codon CD 59 และ CD30 การตรวจ CBC และการหาชนิดฮีโมโกลบิน ไม่สามารถบอกความผิดปกติแบบนี้ได้ ผลการตรวจอาจออกมาปกติแต่ก็ยังสามารถมีภาวะแฝงเหล่านี้ได้ ดังนั้น จึงต้องตรวจระดับยีนเท่านั้น
3. Premium Prevention
- ครอบคลุมชนิดการกลายพันธุ์ที่พบบ่อย ∼99%
- เหมาะสำหรับควบคุมป้องกันโรคธาลัสซีเมียทุกแบบที่พบในประชากรชาวไทย
การตรวจประกอบด้วย
- การตรวจความสมบูรณ์ของเม็ดเลือด CBC
- การหาชนิดและปริมาณ ฮีโมโกลบินด้วยวิธี Capillary Electrophoresis (CE)
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการขาดหายของยีน 7 แบบ ได้แก่ SEA ,THAI, 3.7 , 4.2, MED, FIL , 20.5
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการเปลี่ยนลำดับเบสใน DNA 6 แบบ ได้แก่ฮีโมโกลบิน Constant Spring, ฮีโมโกลบิน Pakse, ฮีโมโกลบิน Quang Sze, การกลายพันธุ์ชนิด initiation codon CD 59 และ CD30
- การตรวจการกลายพันธุ์ชนิดเบต้า ธาลัสซีเมีย อีก 16 แบบโดยวิธี Advanced beta globin ARMS-PCR ได้แก่การกลายพันธุ์ชนิด 28 , CD8/9, CD 17 , CD 19, CD26 (Hb E), CD 26 G>T (stop codon), CD27/28, IVSI-I, IVSI-5, CD 35, CD 41, CD41/42 , CD43, CD 71/72, CD 95 และ IVSII-654)
- การตรวจการขาดหายของยีนเบต้า 10 แบบ ได้แก่ 619bp, 3.5kb, SEA HPFH, Filipino, Hb Lepore, Asian Indian deletions inversion, Chinese, HPFH-6,Thai(δβ)° และ Siriraj-thalassemia
- การตรวจยีนแอลฟ่าโกลบินที่เกินมา (alpha globin triplication) ในรายที่ไม่ได้มีการตรวจระดับยีน การใช้ข้อมูล CBC และ การตรวจชนิดฮีโมโกลบิน อย่างเดียว จะทำให้การวินิจฉัยโรคโลหิตจางธาลัสซีเมียผิดพลาดได้ เนื่องจาก การมีภาวะแฝง alpha globin triplication
4. Complete Diagnostic Package
- ครอบคลุมการกลายพันธุ์ทั้งหมดของโกลบินยีน 100%
- เหมาะสำหรับผู้ที่ต้องการตรวจแบบครอบคลุม100%และวินิจฉัยผู้ป่วยโรคโลหิตจางธาลัสซีเมียหรือผู้ที่ต้องการตรวจแบบละเอียดครอบคลุมมากที่สุด
การตรวจประกอบด้วย
- การตรวจความสมบูรณ์ของเม็ดเลือด CBC
- การหาชนิดและปริมาณ ฮีโมโกลบินด้วยวิธี Capillary Electrophoresis (CE)
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการขาดหายของยีน 7 แบบ ได้แก่ SEA ,THAI, 3.7 , 4.2, MED, FIL , 20.5
- การตรวจการกลายพันธุ์ ของอัลฟ่าธาลัสซีเมียชนิดการเปลี่ยนลำดับเบสใน DNA 6 แบบ ได้แก่ฮีโมโกลบิน Constant Spring, ฮีโมโกลบิน Pakse, ฮีโมโกลบิน Quang Sze, การกลายพันธุ์ชนิด initiation codon CD 59 และ CD30
- การตรวจการกลายพันธุ์ชนิดเบต้า ธาลัสซีเมีย อีก 16 แบบโดยวิธี Advanced beta globin ARMS-PCR ได้แก่การกลายพันธุ์ชนิด 28 , CD8/9, CD 17 , CD 19, CD26 (Hb E), CD 26 G>T (stop codon), CD27/28, IVSI-I, IVSI-5, CD 35, CD 41, CD41/42 , CD43, CD 71/72, CD 95 และ IVSII-654)
- การตรวจการขาดหายของยีนเบต้า 10 แบบ ได้แก่ 619bp, 3.5kb, SEA HPFH, Filipino, Hb Lepore, Asian Indian deletions inversion, Chinese, HPFH-6,Thai(δβ)° และ Siriraj-thalassemia
- การตรวจยีนแอลฟ่าโกลบินที่เกินมา (alpha globin triplication)
- การหาลำดับเบสในยีน alpha globin และ beta globin อย่างละเอียด
หมายเหตุ: เนื่องจากการตรวจวินิจฉัยโรคโลหิตจางธาลัสซีเมียมีความซับซ้อนในการตรวจดังนั้นรายการตรวจที่ 5-8 ใน Premium prevention packageและ Complete diagnostic แพทย์ผู้เชี่ยวชาญจะพิจารณาตามความเหมาะสม
ปัญหาของการตรวจธาลัสซีเมียในห้องปฏิบัติการทั่วไป
โรคธาลัสซีเมียเป็นปัญหาสาธารณสุขที่สำคัญ คนไทยคนไทยมียีนที่เป็นพาหะของโรคประมาณ 25 ล้านคน คนไทยป่วยเป็นโรคธาลัสซีเมียกว่า 6 แสนคน ในแต่ละปีมีเด็กเกิดใหม่ป่วยเป็นโรคธาลัสซีเมีย 25,000 ราย ชนิดรุนแรงและปานกลางเกิดขึ้นเสมอ นโยบายการควบคุมและป้องกันของภาครัฐในปัจจุบันคือการตรวจคัดกรองในหญิงตั้งครรภ์ หากทารกในครรภ์ได้รับการวินิจว่าเป็นโรคธาลัสซีเมียชนิดรุนแรง รัฐอนุญาตให้ทำแท้งได้ นโยบายนี้ยังล้มเหลวและยังมีผู้ป่วยใหม่เกิดขึ้นทุกปีเนื่องจาก
- ห้องปฏิบัติการทั่วไปไม่สามารถตรวจ DNA ได้
- ในปัจจุบันการตรวจคัดกรองในหญิงตั้งครรภ์ จะเริ่มจากการตรวจกรองก่อนไปจนถึงการ ตรวจยืนยันซึ่งใช้เวลานานทำให้ หญิงตั้งครรภ์บางรายที่ทารกในครรภ์ได้รับการวินิจฉัยว่าเป็นโรคตัดสินใจเลือกที่จะตั้งครรภ์ต่อเพราะอายุครรภ์มาก
- นโยบายการตรวจคัดกรองในปัจจุบัน ครอบคลุมเฉพาะโรคธาลัสซีเมียชนิดรุนแรงเท่านั้น ไม่ครอบคลุมการกลายพันธุ์ทุกชนิด ทำให้การแปลผลจำกัด คือไม่สามารถบอกได้ 100 เปอร์เซ็นต์ว่าไม่เป็นภาวะเสี่ยงหรือไม่ และยังพบว่าคู่สมรสได้ตรวจคัดกรองไปแล้วแต่ลูกที่เกิดมาเป็นโรคธาลัสซีเมีย ปัญหานี้พบได้ทั้งใน รพ.เอกชน และรพ.ของรัฐบาล การตรวจวินิจฉัยยีนโรคธาลัสซีเมียมีความซับซ้อนมากทางห้องปฏิบัติการ ต้องมีความรู้และความชำนาญเป็นพิเศษ แพทย์ทั่วไปขาดความรู้ความชำนาญในการอ่านการแปลผล หรือแม้กระทั่งการเลือกตรวจในรายการตรวจที่เหมาะสม
- เทคโนโลยีในการตรวจโรคธาลัสซีเมียมีความซับซ้อนมาก เจ้าหน้าที่ห้องปฏิบัติการต้องมีความรู้และความชำนาญ แพทย์ทั่วไปไม่สามารถอ่านผลหรือแปลผลได้แม้แพทย์จะ ผ่านการฝึกอบรมมาแล้วก็ตาม แพทย์ทั่วไปไม่สามารถเลือกรายการทดสอบที่เหมาะสม เพื่อจะวินิจฉัยหรือให้คำปรึกษาได้
โรคโลหิตจางธาลัสซีเมีย
เนื่องจากอุบัติการณ์ความชุกของโรคธาลัสซีเมียในประเทศไทยถือว่ามากที่สุดประเทศหนึ่งในโลก แม้ว่าปัจจุบันโรคธาลัสซีเมียสามารถรักษาให้หายขาดได้ด้วยการปลูกถ่ายเซลล์ต้นกําเนิด แต่ยังมีข้อจำกัดอยู่มากเนื่องจากการรักษาดังกล่าวมีค่าใช้จ่ายที่สูงและหาผู้บริจาคเซลล์ได้ยากนอกจากนี้ยังมีความเสี่ยงที่จะเป็นอันตรายถึงชีวิตจากการรักษาได้ สิ่งสำคัญที่ประชาชนทั่วไปน่าตระหนักมากที่สุดคือการวินิจฉัยผู้ที่มีภาวะแฝงในสังคมให้ถูกต้องอันจะนำไปสู่การป้องกันโรค เพื่อลดจำนวนผู้ป่วยที่เกิดขึ้นใหม่ด้วยวิธีการที่เหมาะสม
โรคโลหิตจางธาลัสซีเมียเป็นโรคทางพันธุกรรมชนิดพันธุกรรมแบบยีนด้อย หรือ autosomal recessive โรคพันธุกรรมชนิดนี้มีพันธุกรรมที่ผิดปกติในการสร้างโปรตีนโกลบิน(Globin)โดย Globin เป็นโปรตีนสำคัญที่เป็นองค์ประกอบของโปรตีนชนิดหนึ่ง คือ ฮีโมโกลบิน (Hemoglobin) ซึ่ง ฮีโมโกลบินประกอบด้วย 2 ส่วน ส่วนที่เป็น Heme และส่วนที่เป็น Globin ซึ่ง Hemoglobin เป็นโปรตีนที่ทำหน้าที่สำคัญในการขนถ่ายออกซิเจนจากปอดไปยังอวัยวะภายในต่างๆ และเป็นองค์ประกอบสำคัญของเม็ดเลือดแดง และเม็ดเลือดแดงทำหน้าที่ขนถ่ายออกซิเจนได้เพราะมี Hemoglobin อยู่ภายในเมื่อไรก็ตามที่มีความผิดปกติทางพันธุกรรมที่ทำให้สร้างสายโกลบินไม่ได้ จะทำให้โครงสร้างและหน้าที่สร้างของ Hemoglobin ผิดปกติซึ่งนั้นเป็นสาเหตุที่นำไปสู่ภาวะโรคโลหิตจางธาลัสซีเมีย
ผู้ที่มีความผิดปกติของการสร้าง Globin จะมีภาวะซีด ซึ่งภาวะซีดมีระดับความรุนแรงที่แตกต่างกันไปนับตั้งแต่ ซีดตั้งแต่เด็กอยู่ในท้องจนกระทั่งคลอดออกมาหรือเป็นผู้ใหญ่ ภาวะซีดที่เกิดขึ้นมีผลเสียต่อร่างกาย ถ้าซีดมากตั้งแต่อายุน้อยๆ มีผลต่อการเจริญเติบโต ภาวะซีดกระตุ้นให้ร่างกายมีการปรับตัวเพื่อต่อสู้กับภาวะซีด เช่น เวลาเกิดภาวะซีด มีการสร้างเม็ดเลือดได้น้อยลง ร่างกายต้องไปสร้างเม็ดเลือดที่ตำแหน่งอื่นๆนอกเหนือจากที่ไขกระดูกตามปกติ เพราะฉะนั้นผลที่เกิดขึ้นคือ ผู้ป่วยจะมีม้ามโต ตับโต มีก้อนที่อยู่ในกะโหลกศีรษะ มีก้อนอยู่ที่ไขสันหลัง มีก้อนอยู่ในช่องอก เพื่อจะสร้างเม็ดเลือดขึ้นมาใหม่ นอกจากนี้เม็ดเลือดที่ขาด Hemoglobin จะมีอายุที่สั้นลง เพราะฉะนั้นเม็ดเลือดก็จะแตกง่ายขึ้น ผู้ป่วยจะมีอาการตัวเหลือง ตาเหลืองไม่แข็งแรง เมื่อเม็ดเลือดแตกมากๆ จะทำให้เกิดนิ่วในถุงน้ำดี และมีผลเสียในระยะยาวที่ตามมา
เมื่อเกิดภาวะซีด ผู้ป่วยจำเป็นต้องได้รับเลือด ในขณะเดียวกันร่างกายก็จะถูกกระตุ้นให้มีการดูดซึมธาตุเหล็กเพิ่มขึ้นผ่านทางลำไส้เมื่อเวลาผ่านไป ก็จะเกิดภาวะเหล็กเกิน เมื่อเกิดภาวะเหล็กเกินมากขึ้นจนถึงจุดหนึ่งเกินความสามารถที่ร่างกายจะได้รับได้ เหล็กจะเข้าไปสะสมในอวัยวะภายใน เช่น ตับ หัวใจ ตับอ่อน สมอง กระดูก เป็นต้น ทำให้ผู้ป่วยกลายเป็นโรคอื่น ๆ ตามมา เช่น สะสมในตับทำให้เกิดตับแข็ง สะสมในตับอ่อนทำให้เกิดเบาหวาน สะสมในกระดูกทำให้เกิดภาวะกระดูกพรุน สะสมในต่อมใต้สมอง ทำให้ฮอร์โมนไม่หลั่ง เกิดปัญหาทางด้านพัฒนาการทางเพศ สะสมในรังไข่อาจทำให้มีปัญหาเรื่องการสืบพันธุ์และเป็นหมันได้
ชนิดของโรคโลหิตจางธาลัสซีเมียที่สำคัญในประเทศไทย
อัลฟ่าธาลัสซีเมีย เบต้าธาลัสซีเมีย
ผู้ป่วยโรคโลหิตจางธาลัสซีเมีย
โรคโลหิตจางธาลัสซีเมียในประเทศไทย
ภาวะโรคพันธุกรรมชนิดที่มีความผิดปกติของสายโกลบินโปรตีน(Globin) เรียกว่า ธาลัสซีเมีย (Thalassemia) โรคนี้พบมากในแถบเมดิเตอร์เรเนียน ได้แก่ ประเทศอิตาลี ประเทศกรีซ ประเทศตุรกี โมร็อกโก ไซปรัส สำหรับประเทศไทยเป็นประเทศแรกในภูมิภาคเอเชียที่ค้นพบโรคนี้ในแถบเอเชีย เป็นผลงานจากทีมแพทย์ศิริราชที่ค้นพบโรคนี้ตั้งแต่ประมาณหลังสงครามโลกครั้งที่ 2 โดยรายงานพบผู้ป่วยที่มีลักษณะเม็ดเลือดผิดปกติและรายงานเป็นครั้งแรกในภูมิภาคเอเชีย
จากการศึกษาวิจัยที่ทำในประเทศไทยประมาณ 50 ปีแล้ว พบว่าอุบัติการณ์ของคนที่เป็นโรคนี้ที่เป็นพาหะโรคนี้ประมาณ 25-30% ของคนไทย และคนที่เป็นโรคประมาณ 1% ในปัจจุบันจากการศึกษาวิจัยล่าสุดของ รศ.ดร.นพ.วิปร วิประกษิตที่โรงพยาบาลศิริราชเมื่อปีที่แล้ว พบว่าจำนวนคนที่เป็นพาหะโรคนี้ในประเทศไทยมีสูงถึงมากกว่า 40% ฉะนั้นคนที่มีโอกาสที่จะเป็นโรคนี้ในประเทศไทย คิดว่าน่าจะมีไม่ต่ำกว่า 3% ของคนไทยทั้งประเทศ
โดยปกติโรคทางพันธุกรรมมี 2 ชนิด คือ โรคพันธุกรรมเด่น และโรคพันธุกรรมด้อย โรคพันธุกรรมเด่น หมายความว่า เมื่อพ่อหรือแม่ที่เป็นโรคแล้ว โรคนี้ถ่ายทอดมาให้ลูกและหลานต่อไป โอกาสเกิดโรคประมาณ 50% ซึ่งโรคพันธุกรรมเด่นพบไม่ค่อยมาก เพราะผู้ป่วยที่มีโรคพันธุกรรมชนิดนี้มักเสียชีวิตไปก่อน ตั้งแต่อายุยังน้อย สำหรับโรคพันธุกรรมด้อยจะพบมากกว่า ซึ่งโรคทางพันธุกรรมส่วนใหญ่ที่มีในโลกจะเป็นโรคพันธุกรรมด้อย หรือเรียกว่า Autosomal Recessiveโรคโลหิตจางธาลัสซีเมียก็เป็นหนึ่งในโรคพันธุกรรมด้อยที่พบมากที่สุดในโลกและพบมากในภูมิภาคเอเชียตะวันออกเฉียงใต้หรือภูมิภาค AEC อีกด้วย
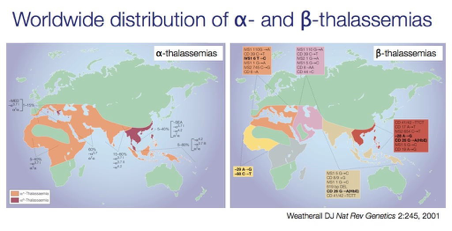
รูปที่ 1. โรคโลหิตจางธาลัสซีเมียก็เป็นหนึ่งในโรคพันธุกรรมด้อยที่พบมากที่สุดในโลก และพบมากในแถบศูนย์สูตร และเมดิเตอร์เรเนียน

รูปที่ 2. ความชุกของโรคโลหิตจางธาลัสซีเมียและโกลบินผิดปกติในภูมิภาคเอเชียตะวันออกเฉียงใต้
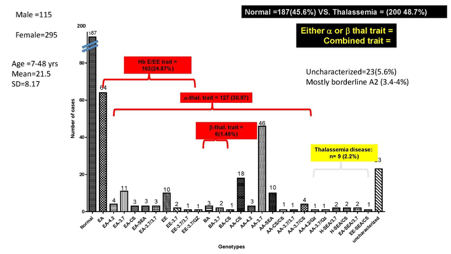
รูปที่ 3. ผลการศึกษายีนโรคโลหิตจางธาลัสซีเมียในประชากรวัยเจริญพันธุ์ (13-45 ปี)
ข้อมูลจากโครงการวิจัยร่วมระหว่างศูนย์ธาลัสซีเมีย คณะแพทยศาสตร์ศิริราชพยาบาล และโรงพยาบาลอ่าวอุดม อ.ศรีราชา จ.ชลบุรี สนับสนุนโดยบริษัทไทยออยล์ จำกัด มหาชน
มนุษย์เรามีโครโมโซม 46 โครโมโซม หรือ 23 คู่ แต่ละโครโมโซมมีข้างหนึ่งที่เราต้องได้มาจากพ่อและอีกข้างได้จากแม่ เพราะฉะนั้นในโครโมโซมที่ทำให้เกิดโรคโลหิตจางธาลัสซีเมียข้างหนึ่งมีพันธุกรรมผิดปกติ อีกข้างหนึ่งยังเป็นพันธุกรรมที่ปกติ เพราะฉะนั้นคนที่มีพันธุกรรมผิดปกติข้างเดียว อีกข้างยังปกติ อยู่ เรียกว่า มีพันธุกรรมแฝงหรือเป็นพาหะ (heterozygote) ซึ่งจะไม่แสดงอาการหรือมีอาการน้อยมาก อาจจะมีอาการเพลีย หรือซีดแต่ไม่รุนแรงและไม่จำเป็นต้องได้รับเลือด ส่วนใหญ่ผู้มีพันธุกรรมแฝงหรือเป็นพาหะ จะไม่ได้รับการวินิจฉัย เพราะการวินิจฉัยค่อนข้างยากต้องใช้ความรู้และเทคโนโลยี ต้องใช้ผู้ที่มีประสบการณ์ ซึ่งปัญหาจะเกิดขึ้นเมื่อคนที่เป็นพาหะกับพาหะมาแต่งงานกัน นำพันธุกรรมที่ผิดปกติทั้ง 2 ข้าง มารวมกัน เด็กที่ออกมาอาจจะมีพันธุกรรมผิดปกติทั้งคู่ จะเกิดเป็นโรคโลหิตจางธาลัสซีเมียขึ้น
โดยทั่วไปโรคโลหิตจางธาลัสซีเมีย แบ่งเป็น 2 ชนิด คือ
- อัลฟาธาลัสซีเมีย (Alpha-thalassemia)
- เบต้าธาลัสซีเมีย (Beta-thalassemia)
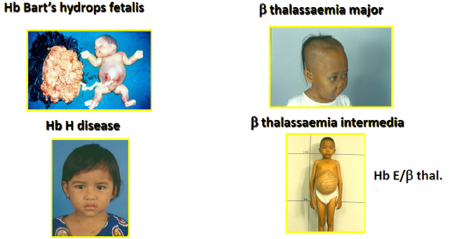
ทั้ง 2 ชนิดมีความรุนแรงที่แตกต่างกันไปตามชนิดย่อยอีกเช่นกัน ชนิด อัลฟาธาลัสซีเมีย (Alpha-thalassemia) มีความรุนแรงตั้งแต่มากที่สุดคือเสียชีวิตจากภาวะซีดตั้งแต่อยู่ในครรภ์เรียกว่า Hb Bart’s Hydrops ไปจนถึงภาวะซีดไม่มาก HbH disease สำหรับชนิดเบต้าธาลัสซีเมีย (Beta-thalassemia) จะซับซ้อนมากกว่า มีทั้งเบต้าธาลัสซีเมียฮีโมโกลบินอี (Beta-Thalassemia/Hemoglobin E) และเบต้าธาลัสซีเมียเมเจอร์ซึ่ง ผู้ป่วยมีอาการรุนแรง มีภาวะซีดมากต้องให้เลือด
อัลฟาธาลัสซีเมีย (Alpha-thalassemia) และเบต้าธาลัสซีเมีย (Beta-thalassemia) แตกต่างกันในแง่ของกลไกการเกิด อาการแสดง ความรุนแรงของโรค และโอกาสในการเกิดภาวะแทรกซ้อน
ต่างๆ เช่น อัลฟาธาลัสซีเมีย ชนิดไฮดรอป์ (Hb Bart’s Hydrops) จะมีการเสียชีวิตตั้งแต่อยู่ในท้อง แต่เบต้าธาลัสซีเมีย จะเริ่มซีดเมื่อคลอดออกมามีชีวิต ภาวะแทรกซ้อนบางอย่างจะพบมากในเบต้าธาลัสซีเมีย เช่น ภาวะความดัน เลือดในปอดสูง เป็นต้น
โดยสรุปโรคโลหิตจางธาลัสซีเมีย เป็นโรคทางพันธุกรรม ผลที่เกิดขึ้นคือภาวะซีด ซึ่งส่งผลต่อร่างกายมากมาย ถ้าเราไม่ดูแลผู้ป่วยอย่างดีตั้งแต่ต้น เมื่อถึงระยะต่อมาผู้ป่วยจะเกิดภาวะแทรกซ้อน เช่น โรคเบาหวาน โรคหัวใจ โรคตับ มีปัญหาเรื่องต่อมไร้ท่อ กระดูกพรุน มีปัญหาเกี่ยวกับทางด้านเพศ ตามมา
การตรวจวินิจฉัยโรคโลหิตจางธาลัสซีเมีย
การตรวจวินิจฉัยโรคธาลัสซีเมียเป็นเรื่องที่มีความสำคัญมาก และเป็นเรื่องที่ต้องเน้นมากสำหรับประเทศไทย เพราะโรคนี้เป็นหนึ่งในโรคทางพันธุกรรมที่เราควบคุมป้องกันได้ ถ้าเราวินิจฉัยโรคได้ก่อนที่จะมีบุตร จะสามารถที่จะลด จำนวนผู้ป่วยที่เกิดใหม่ได้ในแต่ละปี
เนื่องจากประเทศไทย มีคนที่เป็นพันธุกรรมแฝงมากกว่า 40% ถือว่ามากที่สุดแห่งหนึ่งในโลก โอกาสที่คนซึ่งเป็นพันธุกรรมแฝง จะมีพันธุกรรมที่ตรงกันสูงมาก เช่น มีพันธุกรรมแฝงอัลฟาธาลัสซีเมียมาเจอกับแฝงอัลฟาธาลัสซีเมีย หรือมีพันธุกรรมแฝงเบต้าธาลัสซีเมียมาเจอกับเบต้าธาลัสซีเมียเป็นต้น เพราะฉะนั้นเรามีการประมาณการว่าในแต่ละปี ว่ามีเด็กเกิดใหม่ทั่วประเทศไม่ต่ำกว่า 25,000 คน มีโอกาสที่จะเป็นโรคโลหิตจางธาลัสซีเมียหรือประมาณ 3% ของประชากรชาวไทย
ในกรณีของคนที่เป็นพาหะโรคนั้น การวินิจฉัยโรคยากมาก เพราะหน้าตาของพาหะธาลัสซีเมียไม่มีการเปลี่ยนแปลงทางกายภาพ มีวิธีการตรวจอย่างเดียวคือการตรวจเลือด แต่การตรวจเลือดในการตรวจสุขภาพประจำจะไม่มีการตรวจเกี่ยวกับเรื่องนี้โดยตรง ดังนั้นจึงมีความจำเป็นอย่างยิ่งที่จะให้การตรวจธาลัสซีเมียเป็นส่วนหนึ่งของการตรวจสุขภาพด้วย และเรื่องนี้เป็นเรื่องสำคัญของประเทศ เพราะในแต่ละปีรัฐบาลใช้เงินเป็นหมื่นล้านในการรักษาผู้ป่วยโรคธาลัสซีเมีย เพราะฉะนั้นสิ่งที่ดีที่สุดคือการใช้วิธีการควบคุมและป้องกัน
การตรวจคัดกรองตามแนวทางของกระทรวงสาธารณสุขเรื่องการควบคุมโรคธาลัสซีเมีย ที่ได้ทำมาตั้งปี พ.ศ.2532 และตอนนี้นโยบายก็ยังไม่เปลี่ยนแปลงนโยบายนี้จะควบคุมธาลัสซีเมีย 3 ชนิด คือ ชนิดเบต้าธาลัสซีเมียที่รุนแรง ชนิดเบต้าธาลัสซีเมียฮีโมโกลบินอี และชนิดอัลฟาธาลัสซีเมียที่เสียชีวิตในท้อง (ไฮดรอป์)
การตรวจคัดกรองหญิงตั้งครรภ์ ว่ามีพาหะที่จะนำไปสู่โรคทั้ง 3 ชนิดหรือไม่ ซึ่งขั้นตอนในการตรวจมีหลายลำดับขั้นตอน และใช้เวลามาก เมื่อผลออกมาว่าเป็นพาหะ ก็ต้องตามสามีมาตรวจ ซึ่งอัตราที่ตามผู้ชายมาตรวจได้มีประมาณ 30% เท่านั้น ถ้าผู้ชายมาตรวจและผลออกมาว่าเป็นพาหะที่ตรงกัน และลูกมีสิทธิ์เป็นโรคและมีโอกาสรุนแรง ขั้นต่อไปต้องวินิจฉัยลูกในท้อง ต้องนำเลือดจากเด็กในท้องมาตรวจ ถ้าผลออกมาว่าเด็กเป็นโรค แม่สามารถทำแท้งได้โดยไม่ผิดกฎหมาย แต่ปัญหาคือ พ่อแม่ไม่อยากทำแท้ง หรืออายุครรภ์มากเกินกว่าที่จะทำแท้งได้ ผลคือยิ่งมีเด็กเกิดใหม่ทุกปีที่เป็นโรคนี้ และจะกลายเป็นปัญหาของประเทศในระยะยาว
ปัญหาธาลัสซีเมียเป็นปัญหาสำคัญของประเทศไทย ซึ่งในประเทศไทยได้ดำเนินการกว่า 30-40 ปีแล้วแต่ว่าเรายังไม่สามารถแก้ปัญหาได้อย่างถูกต้องเพราะ ยังมีผู้ป่วยใหม่เกิดขึ้นมาเรื่อย ๆ ขึ้นทุกปี ในปี พ.ศ.2556 รศ.ดร.นพ. วิปร วิประกษิต และคณะได้ไปทำโครงการวิจัยที่บ้านอ่าวอุดม อำเภอศรีราชา แหลมฉบัง ซึ่งเป็นโครงการศึกษาวิจัยที่ได้รับทุนจากเครือบริษัทไทยออยล์ เป็นโครงการ ศึกษาวิจัยเพื่อหาพาหะโรคเลือดจางธาลัสซีเมียในชุมชนที่ลงลึกถึงระดับดีเอ็นเอครั้งแรกในไทย โดยตรวจหญิงและชายในชุมชนในวัยเจริญพันธุ์ทุกคนจากการวิจัยดังกล่าวชี้ให้เห็นว่าหาก เราเพิ่มความรับรู้ในประชากร ว่าแต่ละคนใครมีพันธุกรรมผิดปกติอะไรในตัวเอง และหากบุคคลนั้นจะแต่งงานทุกคนควรจะทราบว่าตนเองและคู่สมรสมีพันธุกรรมอะไรที่จะมีผลต่อลูกได้
ส่วนหนึ่งของโครงการที่ทำที่อ่าวอุดมคือ เมื่อคณะผู้วิจัยลงไปตรวจประชากรในหมู่บ้าน ในชุมชน พร้อมกับให้ความรู้ และตรวจธาลัสซีเมียให้ลงลึกในระดับดีเอ็นเอ จากนั้นคณะผู้วิจัยจะมอบผลตรวจกับผู้เข้าร่วมวิจัยทุกคนเอาไว้เป็นข้อมูลส่วนตัว ซึ่งข้อมูลทุกอย่างจะลงรายละเอียดอย่างลึกมาก แล้วแจ้งว่าถ้าคุณเป็นพาหะชนิดไหน จะมีคำแนะนำที่เป็นทางเลือกให้ทั้งหมด ซึ่งทางเลือกมีหลายวิธีด้วยกัน เช่น เมื่อรู้ว่าเป็นพาหะแฝง ก็จะต้องเลือกคู่สมรสที่ไม่เป็นพาหะแฝงชนิดที่ตรงกัน หรือถ้าต้องแต่งงานกับผู้ที่มีพาหะแฝง อาจตัดสินใจไม่มีลูก หรือไปรับเด็กบุญธรรมมาเลี้ยง เป็นต้น หรืออาจใช้อีกทางเลือกที่ทางเราให้บริการอยู่เรียกว่า เทคโนโลยี PGD (Preimplantation Genetic Diagnosis) คือทำการวินิจฉัยโรคทางพันธุกรรมก่อนการฝังตัว โดยการนำเชื้ออสุจิกับไข่มาผสมกันในหลอด ให้เป็นกลุ่มเซลล์ประมาณ 10-20 เซลล์ จากนั้นดึงออกไปตรวจ 1 เซลล์ เพื่อตรวจดูว่ากลุ่มเซลล์นั้นมีพันธุกรรมโรคธาลัสซีเมียหรือไม่ ถ้าตรวจพบก็ไม่นำมาใช้ ถ้าเซลล์ ไหนตรวจแล้วไม่มีพันธุกรรม ก็สามารนำ และเลี้ยงต่อให้เป็นตัวอ่อน จากนั้นนำตัวอ่อนไปฝังตัวในผู้หญิง ซึ่งวิธีการนี้เป็นการลดปัญหาการทำแท้งได้ พ่อแม่ก็ไม่ต้องมีปัญหาในระยะยาว
เทคโนโลยี PGD ในต่างประเทศเป็นที่นิยมมาก ตอนนี้เทคโนโลยีนี้ก้าวหน้าขึ้นไปอีกระดับ นอกจากการตรวจตัวอ่อนเพื่อหาโรคพันธุกรรมธาลัสซีเมียแล้ว ยังสามารถตรวจหาโรคพันธุกรรมชนิดอื่น ๆ ด้วย ซึ่งในอนาคตเป็นเทคโนโลยีที่กำลังจะก้าวต่อไป
ในปัจจุบันซึ่งการรักษาผู้ป่วยธาลัสซีเมียให้หายขาดมีวิธีเดียวคือการปลูกถ่ายสเต็มเซลล์ สเต็มเซลล์ที่เข้ากันได้ดีสำหรับผู้ป่วยก็คือสเต็มเซลล์จากพี่น้องพ่อแม่เดียวกัน การใช้เทคโนโลยี PGDในขณะนี้ไม่ได้ตรวจวินิจฉัยเพียงแค่พันธุกรรมธาลัสซีเมีย เท่านั้น แต่ได้นำมาใช้ในการตรวจกลุ่มเซลล์ว่ามี HLA เข้ากันได้หรือไม่กับผู้ป่วยที่เป็นโรค วัตถุประสงค์คือ ถ้าเด็กคนนี้มีชีวิต คลอดออกมาแล้ว สามารถนำสเต็มเซลล์ไปรักษาผู้ป่วยที่เป็นโรคได้